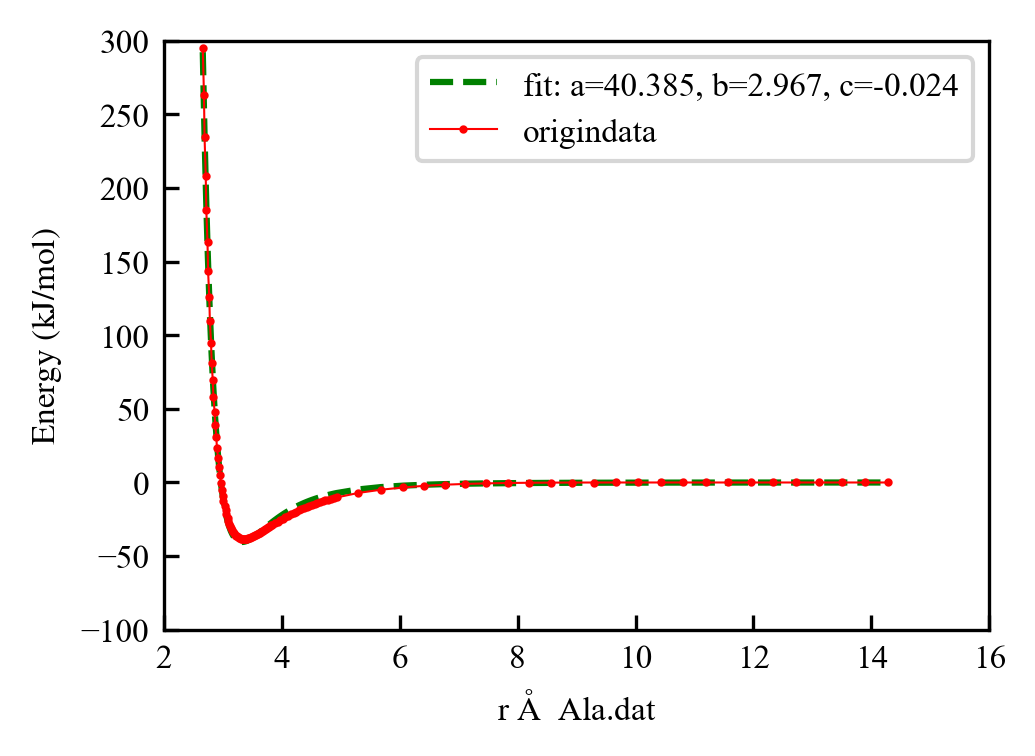
氨基酸粗粒化的Lennard-Jones势函数拟合-shell-python
终于把很久之前想做的给做了, 还是挺麻烦的
整体思路很简单, 单个残基跑md, 然后上下移动残基, 跑em, 得vdw能量与distance, 最后拟合
1. 氨基酸模型与力场
氨基酸使用的是Avogadro1.9程序share库文件夹里自带的单个残基分子构型,文件格式cml
使用obebal程序转化一下格式, 又又又重新记忆了字符串截取, 大小写
# 批量转格式 cml -> pdb
for i in `ls`; do
j=${i:0:-4}
obabel -icml $i -opdb -O ../pdb/$j.pdb
done
# 批量更改名字 eg. Ala.pdb Arg.pdb
for i in `ls`; do
fn=`grep ATOM $i | awk 'END{print $4}'`
a=${fn:0:1}
b=${fn:1}
fn=${a}${b,,}
echo $fn
cp $i ../newname/$fn.pdb
done
再将每个氨基酸重新建个目录存起来, 开始建立力场
设置一下端基不解离,gmx2022 和 gmx2019没啥区别, 该报的bug一样不少
对于几个特殊侧链的氨基酸, 再手动设置一下侧链不解离
for i in `ls`; do
cd $i
echo -e "15\n2\n2\n" | gmx2022 pdb2gmx -water tip3p -ter -ignh -f `basename ${PWD}`.pdb -o `basename ${PWD}`.gro
cd ../
done
# -ter 和 -inter 参数, 手册说明
gmx2022 pdb2gmx -water tip3p -ter -ignh -inter -f `basename ${PWD}`.pdb -o `basename ${PWD}`.gro
2. 建立氧化石墨烯力场
建立一个4x4的片层, 过程就跟之前一样
修改一下topol.top文件
准备开始跑模拟了
3. 氨基酸残基MD模拟
调节好表面文件的位置, box大小, 就直接开始跑
设置em.mdp 和 md.mdp 的能量组; 对于Lys, 比较特殊, 文件不一样, 需要单独的设置
function resmodel {
gmx editconf -f `basename ${PWD}`.gro -o ptw1.gro -box 3.87980 3.78000 3.6
g_adj
gmx editconf -f ptw2.gro -o ptw3.gro -translate 0 0 0.56
g_mer go.gro ptw3.gro
echo -e "a CA CT CF & 13\n name 14 GOC\n q" | gmx make_ndx -f box.gro -o
mkdir em md
}
function resem {
gmx grompp -f em.mdp -c box.gro -p topol.top -o ./em/em.tpr -po ./em/emout.mdp -n
gmx mdrun -v -deffnm ./em/em
}
function resmd {
gmx grompp -f md.mdp -c ./em/em.gro -p topol.top -o ./md/md.tpr -po ./md/mdout.mdp -n -r ./em/em.gro
gmx mdrun -v -deffnm ./md/md -nt 14
}
for i in `ls`; do
cd $i
resmodel
resem
cd ../
done
具体的文件夹设置和目录
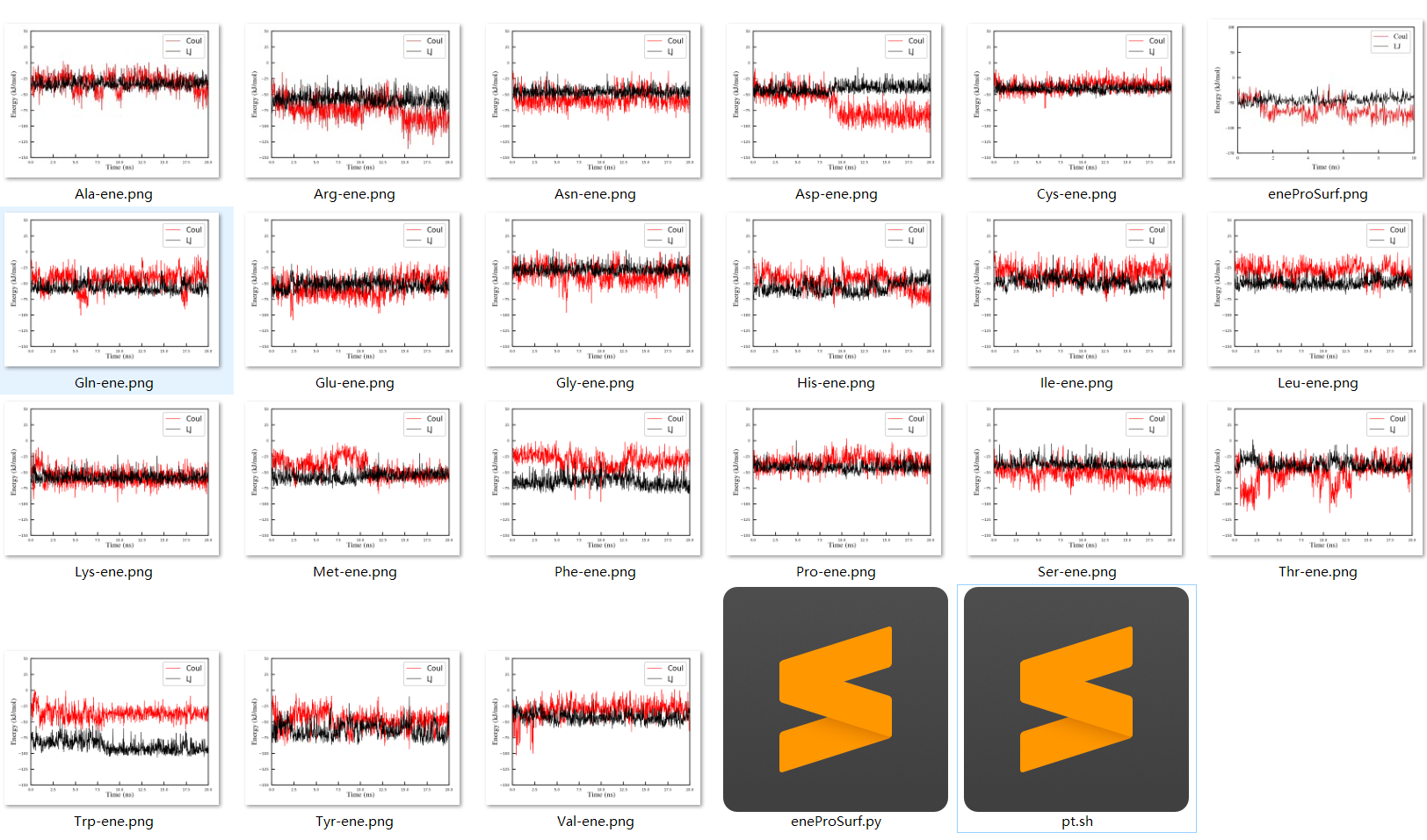
跑完了过后, 瞄瞄能量
resg=(Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys Met Phe Pro Ser Thr Trp Tyr Val)
alias enepy="python ~/ding/gopt/eneSingleRes.py"
for i in `ls`; do
cd $i/md
echo 45 46 | gmx energy -f md.edr -s md.tpr
enepy
cd ../../
cp $i/md/eneProSurf.png ./$i-ene.png
done
看起来应该很稳定
4. 垂直移动氨基酸+em+analysis
一堆坑, 一言难尽, 总之最后踩完了坑, 批处理就很简洁了
设置移动梯度, 固定System, 分析能量和距离
每一个梯度都放在./fit/文件夹下面, 很多但不至于太乱
eg ~/gopt/allres/Ala/fit/fit_0.02/fit.gro
~/gopt/allres/Arg/fit/fit_0.04/fit.gro
Lys需要单独判断处理一下
resg=(Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys Met Phe Pro Ser Thr Trp Tyr Val)
# 导出能量和距离
function edist {
d=`awk 'END{print 10*$NF}' dist.xvg`
v=`awk 'END{print $NF}' energy.xvg`
echo $d $v
}
# 设置数据梯度
grads=($(seq -0.64 0.02 0.8) $(seq 0.85 0.05 2) $(seq 2.4 0.4 12))
function restrans {
for i in ${grads[@]}; do
mkdir -p ./fit/fit_$i
awk -v i="$i" '{
if ($0 ~ / {4}1[A-Z]{3}/) {
$6 += 0.1*i;
printf("%8s%7s%5d%8.3f%8.3f%8.3f\n",$1,$2,$3,$4,$5,$6);
} else {print $0}
}' ./md/md.gro > ./fit/fit_$i/fit.gro
done
}
function fitem {
for i in ${grads[@]}; do
i="fit_$i"
gmx grompp -f fitem.mdp -c ./fit/$i/fit.gro -p topol.top -o ./fit/$i/fit.tpr -po ./fit/$i/fitout.mdp -n
gmx mdrun -v -deffnm ./fit/$i/fit
done
}
function vddat {
rm vdwdist.dat
for i in ${grads[@]}; do
i="fit_$i"
cd ./fit/$i
echo 38 39 | gmx energy -f fit.edr -s fit.tpr
if [[ $wd != Lys ]]; then
gmx pairdist -s fit.tpr -f fit.trr -tu ns -ref "group 13" -sel "com of group 1"
else
gmx pairdist -s fit.tpr -f fit.trr -tu ns -ref "group 3" -sel "com of group 2"
fi
edist >> ../../vdwdist.dat
cd ../../
done
}
function sortdat {
sort -n -k 1 vdwdist.dat > ../`basename $PWD`.dat
}
for wd in ${resg[@]}; do
cd $wd
# restrans
# fitem
# vddat
sortdat
cd ../
done
最终结果加拟合