氧化石墨烯表面力场构建 opls
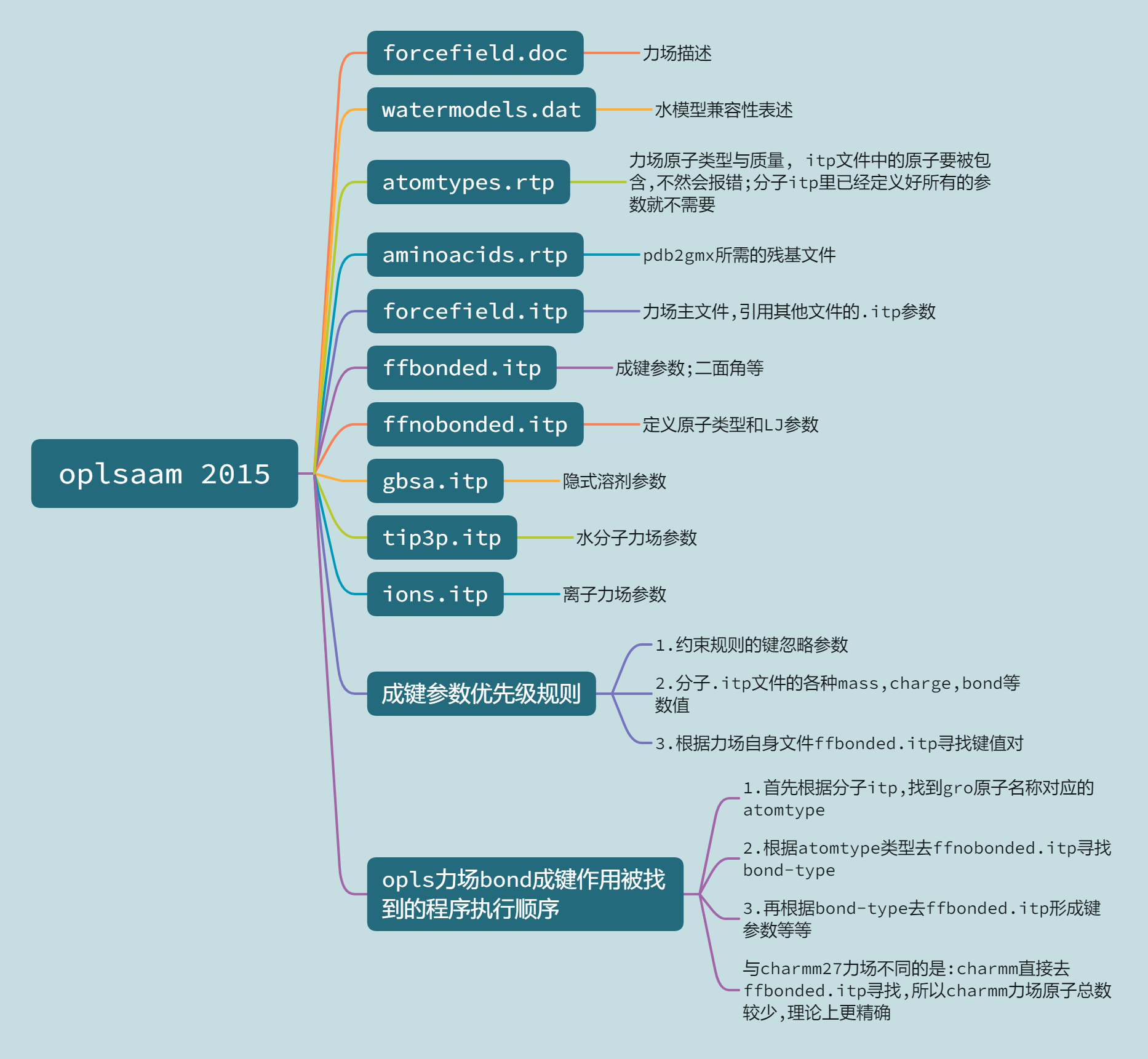
1. oplsaam 2015力场构成
Robertson M J, Tirado-Rives J, Jorgensen W L.Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field[J].Journal of Chemical Theory and Computation,2015, 11 (7): 3499-3509.
2. FAD力场生成
LigParGen程序自动生成, 搞出来FAD的单体 fad_full.pdb
ligpargen -i fad_full.pdb -n fad -p fad.ff -r FAD -c -2 -cgen CM1A-LBCC
生成出来的fad.gmx.itp:
[ atomtypes ]
opls_800 O 15.9990 0.000 A 2.96000E-01 8.78640E-01
opls_801 P 30.9740 0.000 A 3.74000E-01 8.36800E-01
opls_802 O 15.9990 0.000 A 2.96000E-01 8.78640E-01
opls_803 OS 15.9990 0.000 A 2.90000E-01 5.85760E-01
...
[ moleculetype ]
; Name nrexcl
FAD 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass
1 opls_801 1 FAD PO1 1 2.8236 30.9740
2 opls_802 1 FAD O1 1 -0.9868 15.9990
3 opls_800 1 FAD O15 1 -0.9868 15.9990
4 opls_803 1 FAD O2 1 -0.8361 15.9990
...
原子电荷使用的charmm27力场文献所提供的, 也就是第一个体系里面搞的
后面在考虑要不要和Go表面的itp文件一起合并, 新的atomtype只能在一个文件里产生
3. 氧化石墨烯力场生成
大的来了, 应该分几步走
- Jerkwin 在线生成石墨烯, 氧化石墨烯
- 脚本或者MS, MS应该更好, 把文件里原子名改掉, 生成带有成键的mol2文件
- 做成一个大残基放在aminoacids.rtp文件里, 把文献力场参数导进去
- gmx pdb2gmx生成itp文件
- 使用脚本排错
3.1 氧化石墨烯

A: Epoxy bridges, B: Hydroxyl groups, C: Pairwise carboxyl groups.
CA, SP2 carbon 石墨烯
CT, C in epoxy 环氧基
OS, O in epoxy
CF, C bonded with phenolic hydroxy 羟基
OH, O in phenolic hydroxy
HO, H in phenolic hydroxy
C, C in carboxyl 羧基
O_3, =O in carboxyl 羧基双键氧
OH2, O of -0H in carboxyl
HO2, H of -OH in carcboxy1
有两种, 就选择没有羧基的吧.
创建一个10x10nm的氧化石墨烯gro, 转化成pdb
总共5244个原子, 文件名go.gro
3.2 Materials Studio 野路子改名
失败 defeat 野路子终究是野路子
- 计算成键
- 分配力场, 删除不正常识别的成键
- 选区某种原子, 改名, 保存成mol2格式 go.mol2
- 保证不会因为某些原子距离太近, 而被识别成错误的成键
3.3 半正经路子 Python成键判断改名
或者使用Python判断原子类型, 但是成键mol2文件还是需要ms
distJudge.py 根据网页生成的石墨烯有特定的原子排序, 计算成键距离判断原子的种类
- a: 最后一个石墨烯平面C原子序号
- b: 最后一个环氧基O原子序号
- c: 最后一个羟基OH的H原子序号
- d: 最后一个羧基H序号, 也是最后一个原子序号
- awk 是shell用来生成itp的, 现在用不上了
- go.gro生成go.dat, 把go.dat搞成go.gro 原有go改名go-origin
import numpy as np
import os
date = np.genfromtxt('go.gro', dtype=('<U5', '<U5', int, float, float,
float), skip_header=2, skip_footer=1)
a = 4032
b = 4436
c = 5244
d = 5244
date['f0'] = "1OGO"
atomtype = date['f1']
atomnum = date['f2']
x = date['f3']
y = date['f4']
z = date['f5']
for i in range(a):
atomtype[i] = "CA"
for j in range(a,b):
dist_2 = (x[i]-x[j])**2 + (y[i]-y[j])**2 + (z[i]-z[j])**2
dist = dist_2**0.5
if dist < 0.14:
# print(dist,i,j)
atomtype[i] = "CT"
atomtype[j] = "OS"
for j in range(b,c,2):
dist_2 = (x[i]-x[j])**2 + (y[i]-y[j])**2 + (z[i]-z[j])**2
dist = dist_2**0.5
if dist < 0.15:
# print(dist,i,j)
atomtype[i] = "CF"
atomtype[j] = "OH"
atomtype[j+1] = "HO"
for j in range(c,d,4):
dist_2 = (x[i]-x[j])**2 + (y[i]-y[j])**2 + (z[i]-z[j])**2
dist = dist_2**0.5
if dist < 0.16:
# print(dist,i,j)
# atomtype[i] = "CF"
atomtype[j] = "C1"
atomtype[j+1] = "O3"
atomtype[j+2] = "O2"
atomtype[j+3] = "H4"
pass
np.savetxt('go.dat', date, fmt=['%8s', '%7s', '%5d', '%8.3f', '%8.3f', '%8.3f'], delimiter='')
# awk 'BEGIN {i = 0}; {i = i + 1; \
# if ($2 == "CA") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_ca", 1, "OGO", $2, 1, 0); \
# if ($2 == "CT") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_ct", 1, "OGO", $2, 1, 0.14); \
# if ($2 == "OS") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_os", 1, "OGO", $2, 1, -0.28); \
# if ($2 == "CF") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_cf", 1, "OGO", $2, 1, 0.15); \
# if ($2 == "OH") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_oh", 1, "OGO", $2, 1, -0.585); \
# if ($2 == "HO") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_ho", 1, "OGO", $2, 1, 0.435); \
# if ($2 == "C1") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_c1", 1, "OGO", $2, 1, 0.52); \
# if ($2 == "O2") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_o2", 1, "OGO", $2, 1, -0.53); \
# if ($2 == "O3") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_o3", 1, "OGO", $2, 1, -0.44); \
# if ($2 == "H4") printf("%5d%11s%6d%10s%8s%5s%10.3f\n", i, "go_h4", 1, "OGO", $2, 1, 0.45); \
# }' go_0.gro > tt
3.4 pdb2gmx
将石墨烯搞成一个大残基的形式写入aminoacids.rtp, pdb2gmx
- 首先决定好原子的力场类型
; atomtypes.atp
; OGO atomtypes
go_ca 12.01100 ; SP2 carbon
go_ct 12.01100 ; C in epoxy
go_os 15.99940 ; C in epoxy
go_cf 12.01100 ; C bonded with phenolic hydroxy
go_oh 15.99940 ; O in phenolic hydroxy
go_ho 1.00800 ; H in phenolic hydroxy
go_c1 12.01100 ; C in carboxyl
go_o2 15.99940 ; O of -OH in carboxyl
go_o3 15.99940 ; =O in carboxyl
go_h4 1.00800 ; H of -OH in carboxyl
- 先将gro文件每个原子改名, 因为rtp里面原子名称最多四个字符, 且不能重复; group分组代表该组电荷量为0,每一组不超过32个原子;
- 最终结果如下
[ OGO ]
[ atoms ]
A001 go_ca 0.000 1
A002 go_ca 0.000 1
A003 go_ca 0.000 1
...
[ bonds ]
A001 A002
A002 A005
A002 A003
...
操作过程
# 先用ABCDEF0-999 造出来不重复的原子name, 选5244个
# 数组赋值A-Z
markChar=({A..Z})
# 输出标记后的每个原子名, 数字补零
for i in {1..5244}; do
let j=i/1000
let k=i%1000
printf "%c%03d\n" ${markChar[$j]} $k
done > atomMarkName
# 过时方法
# {
# for (( i = 1; i <= 999; i++ )); do echo A$i; done
# for (( i = 0; i <= 999; i++ )); do printf "B%3d\n" $i; done
# for (( i = 0; i <= 999; i++ )); do printf "C%3d\n" $i; done
# for (( i = 0; i <= 999; i++ )); do printf "D%3d\n" $i; done
# for (( i = 0; i <= 999; i++ )); do printf "E%3d\n" $i; done
# for (( i = 0; i <= 999; i++ )); do printf "F%3d\n" $i; done
# } | head -5244 | sed 's/ /0/g' > 1
# 再把gro文件原子名称对应的力场类型搞定, 加上charge电荷量; j = i/30 表示: 30个原子一组,
awk 'BEGIN {i = 0}; {i++; \
j = (i-1)/30 + 1;
if ($2 == "CA") printf("%7s%8.3f%7d\n", "go_ca", 0, j); \
if ($2 == "CT") printf("%7s%8.3f%7d\n", "go_ct", 0.14, j); \
if ($2 == "OS") printf("%7s%8.3f%7d\n", "go_os", -0.28, j); \
if ($2 == "CF") printf("%7s%8.3f%7d\n", "go_cf", 0.15, j); \
if ($2 == "OH") printf("%7s%8.3f%7d\n", "go_oh", -0.585, j); \
if ($2 == "HO") printf("%7s%8.3f%7d\n", "go_ho", 0.435, j); \
}' go.gro > atomResData
# 整合到一块 就是 [ atoms ]区域参数了
echo -e "[ OGO ]\n [ atoms ]" >> aminoacids.rtp
paste -d " " atomMarkName atomResData >> aminoacids.rtp
- 需要使用mol2格式里面的bond成键信息, 转换成键信息为自己所需的
# 提取bond信息,再删除第一行与最后一行
sed -n '/@<TRIPOS>BOND/,/@<TRIPOS>/p' go.mol2 | sed '1d; $d' > goBond
# 65是A的ascii码值, int()是awk内置取整的函数, 也是为了修改键序号转换成相应的atomMarkName
awk '{
printf("%c%03d %c%03d\n", 65+int($2/1000), $2%1000, 65+int($3/1000), $3%1000)
}' goBond > goBandMarkName
# 将成键信息写入rtp
echo " [ bonds ]" >> aminoacids.rtp
cat goBandMarkName >> aminoacids.rtp
- 再将go.gro 中第二列的name全替换成ABCD0-999这种名字,按顺序, 列操作就用编辑器工具吧, 实在是不想写命令了, 存为go-new.gro
- 没办法搞错了,我又重来了一遍, 而且还没有工具人帮我, 还是写个命令吧
head -2 go.gro > go-new.gro
sed '1,2d;$d' go.gro | awk '{ printf("%8s%4c%03d%5d%8.3f%8.3f%8.3f\n",$1,65+int($3/1000),$3%1000,$3,$4,$5,$6)}' >> go-new.gro
tail -1 go.gro >> go-new.gro
GoSurface
5244
1OGO A001 1 0.061 0.070 0.000
1OGO A002 2 0.182 0.140 0.000
1OGO A003 3 0.182 0.280 0.000
... ...
1OGO F242 5242 2.364 4.432 -0.176
1OGO F243 5243 8.426 2.240 0.146
1OGO F244 5244 8.347 2.286 0.176
10.184459 10.080000 0.280000
gmx pdb2gmx -f go-new.gro -o -water tip3p最后一步
3.5 grompp生成力场, 改错
# 按照opls识别规律, 先把atomtypes写入到ffnobonded.itp 为了独一无二我在type字段里都加了一个_*
[ atomtypes ]
go_ca CA_* 6 12.0110 0.000 A 3.55000e-01 2.92880e-01
go_ct CT_* 6 12.0110 0.140 A 3.50000e-01 2.76144e-01
go_os OS_* 8 15.9994 -0.280 A 2.90000e-01 5.85760e-01
go_cf CF_* 6 12.0110 0.150 A 3.55000e-01 2.92880e-01
go_oh OH_* 8 15.9994 -0.585 A 3.07000e-01 7.11280e-01
go_ho HO_* 1 1.0080 0.435 A 0.00000e+00 0.00000e+00
...
# 再把键,角信息写入ffbonded.itp
[ bondtypes ]
CA_* CA_* 1 0.14 392459.2
CA_* CT_* 1 0.151 265265.6
...
[ angletypes ]
CA_* CA_* CA_* 1 120 527.184
CT_* OS_* CT_* 1 109.5 502.08
CA_* CA_* CT_* 1 120 585.76
...
[ dihedraltypes ]
CA_* CA_* CA_* CA_* 3 30.334 0 -30.334 0 0 0
CA_* CT_* OS_* CT_* 3 1.71544 2.84512 1.046 -5.60656 0 0
CA_* CA_* CT_* OS_* 3 0 0 0 0 0 0
...
gmx grompp -f em.mdp -c go-new.gro -p topol.top -o em.tpr nohup 搞一下把报错地方找出来, 缺少的bonds等等参数一个个加上去
- 处理错误数据
rm nohup.out
nohup bash gmx.sh &
sleep 5s
# 提取error里有用的信息, sed 圆括号匹配() 还有[]都需要转义, ()匹配的字符按照顺序存为\1 \2 \3
# ERROR 7149 [file topol.top, line 86843]: 替换成 ERROR 7149 file topol.top, line 86843
sed -n 's|\(^ERROR.*\)\[\(.*line.*\)\]:|\1\2|gp' nohup.out > errlines
# 将报错的top文件行号保存字符串
errLinesStrings=$(awk '{print $6}' errlines)
# 寻找到报错行的键,角队列
awk -v elStr="$errLinesStrings" '
BEGIN {
# 分割字符串成为数组el
split(elStr, el);
i = 1;
} {
if (NR == el[i]) {
i++;
for (j = 1; j < NF; j++) {printf("%8d", $j)}
printf("\n");
}
}
' topol.top > errData
# 将原子存入数组,索引为序号,值为type类型, 0索引不用
# 29就是原子类型开始的前一行
atomType=($(awk 'NR==29,NR==5274 {print $2}' topol.top))
# 将每一行的数据读出来-1, 当做索引, 获取值type, 打印出来
cat errData | while read line; do
for i in $line; do
printf "%8s" ${atomType[$i]}
done
echo
done > errType
# 排序后去重, 所留下来的就是力场文件里需要补充的参数,命令随意符合自己要求就行
sort -u -k 1 -k 2 -k 3 -k 4 errType | tr a-z A-Z > missType
# 力场里没有CF, 小秦说力场里CT就是CF
sed -i 's/GO_//g' missType
3.6 寻找合适参数
问大佬们, 或者找找力场类似的, 或者使用ligpargen试试看有没有类似生成的, 或者直接全部设置成0;
- 最终全部搞定, 把原子名MarkName改回来
atomTureNameStrings=$(sed '1,2d;$d' go.gro | awk '{print $2}')
# 反人类的awk索引从1开始
awk -v anStr="$atomTureNameStrings" 'BEGIN {
split(anStr, an);
} {
i = NR - 29;
if (i > 0 && i <= 5244) { $5 = an[i] }
print $0;
}' topol.top > ogo.itp
3.7 来个em
gmx make_ndx -f box.gro <
a CT CA CF & "OGO"
q
gmx grompp -f em.mdp -c box.gro -p topol.top -o ./em/em.tpr -po ./em/emout.mdp -n
gmx mdrun -v -deffnm ./em/em
搞定!